

Нейрофиброматозы – наследственные заболевания, характеризующиеся образованием доброкачественных опухолей в коже, мягких тканях, нервной системе и внутренних органах. Выделяют 6 типов нейрофиброматозов, клинически значимы типы I и II. Общие симптомы включают нейрофибромы на коже, опухоли спинномозговых корешков, слуховых и зрительных нервов, пигментные пятна, костные деформации. Диагностика основана на данных осмотра пациентов, выявлении опухолей с помощью МРТ и КТ спинного и головного мозга, внутренних органов. Лечение симптоматическое – проводится резекция опухолей, рентгенотерапия, химиотерапия.
- Причины нейрофиброматозов
- Патогенез
- Симптомы нейрофиброматозов
- Осложнения
- Диагностика
- Лечение нейрофиброматозов
- Прогноз и профилактика
- Цены на лечение
Общие сведения
Нейрофибромы – доброкачественные опухоли, развивающиеся из оболочек нервных волокон. Чаще всего располагаются в слоях кожи и подкожной клетчатке, иногда поражают головной мозг, нервные волокна, корешки спинного мозга, мягкие ткани, внутренние органы. Нейрофиброматоз – болезнь, при которой образуются многочисленные нейрофибромы. Распространенность разных типов патологии значительно колеблется: заболеваемость 1 типом составляет 1:2 500, 2 типом – 1:50 000. Другие варианты встречаются еще реже, их точная эпидемиология не определена. Гендерной и расовой предрасположенности не выявлено. Дебют клинических проявлений возможен в любом возрасте, зависит от типа болезни.
Причины нейрофиброматозов
Образование множественных нейрофибром детерминировано генетически. При нейрофиброматозе I существует мутация гена НФ1, расположенного на длинном плече 17 хромосомы. Он относится к генам-супрессорам роста опухолевых тканей, большая часть из которых – нейроэктодермального генеза. При дефекте в гене НФ1 нарушается синтез белков, ответственных за клеточную пролиферацию. Мутации носят характер транслокаций, делеций, инверсий, точковых изменений. Больше 80% из них приводят к синтезу нефункциональных белков или к полному отсутствию белковых молекул. Наследование происходит по аутосомно-доминантному механизму с высокой степенью пенетрации: при наличии мутационного гена у одного из родителей вероятность болезни у ребенка составляет 50%, если оба родителя имеют мутацию, риск повышается до 80-90%. Известны случаи спонтанных мутаций.
Причиной нейрофиброматоза II является мутационное изменение гена НФ2, локализованного на 22 хромосоме. Он кодирует производство белка мерлина (шванномина) – супрессора опухолевого роста. Тип наследования – аутосомно-доминантный с небольшой степенью пенетрации. Передача одного мутантного гена зачастую не проявляется, поскольку второй ген обеспечивает синтез достаточного количества белков. Если он повреждается, синтез нормальных фракций мерлина прекращается, пролиферация клеток усиливается, развивается новообразование. При других типах нейрофиброматозов также существуют мутации в генах, обеспечивающих воспроизведение молекул белков-супрессоров роста опухолей.
Патогенез
Общим патогенетическим механизмом развития нейрофиброматозов является наследственно обусловленная недостаточность какого-либо белка, подавляющего процессы опухолевой пролиферации клеток в тканях нейроэктодермального происхождения. При мутации одного гена производство опухолевых супрессоров прекращается наполовину, равновесие роста и гибели клеток смещается в сторону митотического деления. Нормальный аллельный ген частично компенсирует дефицит белка. Тяжесть нейрофиброматоза определяется тем, насколько дефектный ген влияет на активность белка-супрессора – частично или полностью нарушает функциональность, полностью блокирует производство. Кроме этого, выраженность клинических признаков зависит от сохранности противоопухолевого иммунитета.
Во многих органах и тканях пациентов формируются доброкачественные опухолевые образования, состоящие из соединительной ткани и пигментных клеток. На нервных стволах образуются невриномы; на поверхности кожи – пигментированные области, жировые бляшки, расширенные сосуды; на сетчатке глаз – факоматоз. Изменяется строение костей, они остаются недоразвитыми либо чрезмерно утолщаются, искривляется позвоночный столб.
Симптомы нейрофиброматозов
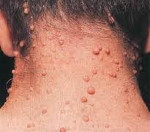
Заболевания проявляются признаками поражения кожи, нервной системы. Классическим клиническим вариантом является нейрофиброматоз типа I, на долю которого приходится 90% случаев болезни. Характерный симптом – гиперпигментация. У больных при рождении или в раннем детстве появляются кожные пятна, цвет которых варьируется от светло-золотистого до коричневого «кофе с молоком». В отдельных случаях пятна имеют фиолетовый или синий оттенок. На радужке глаза обнаруживаются узелки Лиша (пятна пигмента – гамартомы) небольшого размера, белесоватые или светло-бежевые, заметные только при офтальмологическом осмотре. Являются специфическим признаком нейрофиброматоза 1, образуются по мере взросления: у детей до 4 лет распространенность составляет 22%, с 5 до 9 лет – 41%, с 10 до 19 лет – 85%, после 20 лет – 95%.
В период пубертата и позже формируются кожные и плексиформные нейрофибромы, располагающиеся соответственно подкожно (на мелких нервных волокнах, иннервирующих кожу) и на крупных нервах. Они представляют собой небольшие доброкачественные новообразования. Кожные нейрофибромы воспринимаются как косметический дефект, при определенном расположении травмируются. Плексиформные опухоли, локализующиеся по ходу периферических нервов, выявляются на конъюнктиве, веках, в брюшной полости и средостении. Проявляются хронической болью, онемением, судорогами, параличом. Опухоли ЦНС находятся внутри черепа, представлены глиомами зрительных нервных волокон, астроцитомами, эпендимомами, невриномами слухового нерва, менингиомами и нейрофибромами. Клиническая картина определяется размерами новообразований, вовлеченностью мозговых структур в патологический процесс. В детском возрасте диагностируются расстройства психического развития: снижение когнитивных способностей, гиперактивность, редко – деменция.
При тяжелом нейрофиброматозе деформируется костная система. У больных возникает сколиоз, краевые структурные изменения тел позвонков и их отростков, эрозийные поражения краев межпозвоночных отверстий и задних ребер. Характерна атрофия либо, наоборот, гипертрофия трубчатых костей. Кости часто искривлены, на поверхности обнаруживаются периостальные гребни и наслоения. В полостях костей могут образовываться нейрофибромы. Если в процесс вовлекаются кости черепа, появляется внешняя асимметрия, наиболее выраженная при поражении лицевой части и глазниц. Свод черепа может иметь атрофированные участки, дефекты, узуры, иногда отмечается локальное увеличение костного вещества.
При типе 2 формируются высокодифференцированные опухоли, которые, однако, более агрессивны, чем при заболевании 1 типа. Пигментных пятен нет. Образуются невриномы – подвижные и болезненные неоплазии. Нередко они локализуются на слуховом нерве, вызывая потерю слуха. Нейрофиброматоз 3 типа отличается большим количеством нейрофибром, ускоренным развитием нейролемм и глиом зрительного нерва, приводящих к расстройству зрения. Специфический признак – появление нейрофибром на ладонях. При болезни 4 типа симптомы похожи, сохраняется риск поражения зрительных волокон. Для 5 типа характерны пигментные темные пятна, опухоли больших размеров, провоцирующие асимметрию тела. Течение 6 типа сопровождается лишь пигментными пятнами. При 7 типе выявляются нейрофибромы средних размеров, гиперпигментации нет.
Осложнения
В 10% случаев нейрофибромы трансформируются в злокачественные опухоли. В группе высокого риска находятся пациенты с большим катамнестическим стажем, беременные женщины. У 6% детей нарушается умственное развитие: они имеют проблемы при освоении учебных навыков (чтение, письмо, счет), с трудом запоминают новую информацию, долго адаптируются в незнакомых ситуациях. Больные всех возрастов подвержены депрессии, поскольку испытывают дискомфорт, чувство стыда и неловкости из-за обезображенной внешности. Множественные нейрофибромы провоцируют эндокринные расстройства, эпилептические припадки, гипотонию мышц, стеноз почечной и легочной артерии, легочные кисты, интерстициальную пневмонию, гипертрофию клитора, нарушения развития органов ЖКТ.
Диагностика
Подозрение на нейрофиброматоз возникает при множественных подкожных опухолях, пигментных пятнах, спинальной шванноме, ухудшении слуха и зрения. Обследование проводят дерматовенеролог, невролог, офтальмолог, отоларинголог и генетик. Перед инструментальными и лабораторными процедурами осуществляется сбор семейного и личного анамнеза, клинический опрос и осмотр. В ходе генеалогического анализа выявляется передача заболевания в нескольких поколениях, реже определяется первичная спонтанная мутация. На теле пациентов обнаруживаются нейрофибромы, пигментные области (при определенных типах болезни), искривления позвоночника, деформации костей, нарушения зрения, слуха, координации движений. Производится дифференциальная диагностика различных вариантов нейрофиброматозов, исключается синдром Протея, рассеянный липоматоз, синдром Клиппеля-Треноне-Вебера. Для уточнения диагноза назначаются:
- МРТ, КТ. Визуализационные методы исследований позволяют определить наличие нейрофибром в головном мозге, позвоночнике, внутренних органах. Двусторонние невриномы 7 пары черепных нервов являются диагностическим критерием нейрофиброматоза II типа. Часто выявляются глиомы, шванномы, менингиомы. Для I типа свойственно развитие плексиформных и обычных новообразований, глиом.
- Рентгенография костей скелета. Диагностическая процедура выполняется с целью подтверждения и оценки тяжести сколиоза, костных атрофий и гипертрофий, локальных утолщений и эрозийных поражений костных структур. При большинстве типов болезни наблюдается истончение кортикального слоя, ложные суставы, дисплазии крыльев клиновидной кости, дугообразное искривление большеберцовой и малоберцовой костей, кисты длинных костей.
- Офтальмологическое исследование. Нейрофиброматоз типа 1 сопровождается плексиформной нейрофибромой век, меланоцитарными гамартомами радужки, глиомой оптических нервных волокон, астроцитарной гамартомой сетчатки, утолщением роговичных нервов, конъюнктивальной нейрофибромой, ишемическими поражениями венул сетчатки. Патогномоничный признак – пятна светлого оттенка на глазном дне и радужке (гамартомы). При 2 типе диагностируется задняя субкапсулярная катаракта, помутнение хрусталика.
- Аудиологическое исследование. При опухолевом поражении слуховых нервов и жалобах на нарастающую глухоту (тугоухость) выполняется электрокохлеография и импедансометрия. Результаты указывают на снижение остроты слуха, наличие слуховой нейропатии, определяют причину и локализацию нарушения.
Лечение нейрофиброматозов
В настоящее время терапия данной группы заболеваний заключается в симптоматической помощи больным. Пациенты регулярно проходят обследования, нацеленные на контроль формирования и увеличения опухолей. При наличии нейрофибром, провоцирующих боль, расположенных в местах повышенного риска травмирования, сдавливающих или смещающих жизненно важные органы, проводится их хирургическое удаление. Применяются классические методики резекции неоплазий и участков нервов, криодеструкция, лазерная хирургия. При множественных новообразованиях назначается лучевая терапия, химиотерапия. Больным с поражением опорно-двигательного аппарата показаны реабилитационные мероприятия (физиолечение, ЛФК).
Активно разрабатываются способы этиологического лечения нейрофиброматозов. На стадии клинических испытаний находится терапия ингибиторами RAS (белков-активаторов роста опухолей) у лиц с нейрофиброматозом первого типа. Этап теоретических разработок проходят методы генной инженерии. Усилия ученых-генетиков направлены на создание и внедрение в организм больных нормального НФ1 гена, отвечающего за синтез нейрофибромина, на расшифровку и введение гена ФН2, обеспечивающего транскрипцию белка шванномина. В некоторых медицинских центрах предпринимаются попытки применения патогенетической терапии, в основе которой лежит комплексное использование стабилизаторов мембран тучных клеток, антипролиферативных препаратов и ферментов, корректирующих метаболические процессы.
Прогноз и профилактика
Нейрофиброматозы являются прогностически благоприятными заболеваниями – малигнизация опухолей происходит редко, в большинстве случаев больные остаются трудоспособными и социально адаптированными. При правильных и регулярных реабилитационных мероприятиях нарушения со стороны костной системы и задержка умственного развития успешно корректируются. Поскольку заболевание является наследственным, профилактика возможна на этапе планирования беременности, парам из групп риска (с отягощенным семейным анамнезом) рекомендуется медико-генетическое консультирование с определением вероятности рождения больного ребенка.
Источник: https://www.krasotaimedicina.ru/diseases/genetic/neurofibromatosis





