

Синдром Хантера – наследственное заболевание обмена веществ с Х-сцепленным рецессивным типом наследования, характеризующееся дефицитом лизосомального фермента идуронат-2-сульфатазы и накоплением мукополисахаридов в тканях. При синдроме Хантера отмечается задержка роста, макроцефалия, деформация костно-суставного аппарата, поражение кожи, сердечно-сосудистой и дыхательной системы, гепатоспленомегалия, нарушение слуха, умственная отсталость. С целью диагностики синдрома Хантера проводится консультация генетика, определение экскреции гликозаминогликанов, рентгенография костей и суставов. Пациентам с синдромом Хантера показана пожизненная ферментозамещающая терапия препаратом элапраза.

Общие сведения
Синдром Хантера (мукополисахаридоз II типа) – редкое генетическое заболевание, сцепленное с Х-хромосомой, при котором вследствие ферментной недостаточности происходит неполное разрушение и накопление кислых мукополисахаридов (гликозаминогликанов) в различных тканях. Частота рождения детей с синдромом Хантера составляет примерно 1:100000-150000 новорожденных. В настоящее время в мире насчитывается не более 2000 больных синдром Хантера; по России официальная статистика отсутствует. Синдром Хантера относится к группе орфанных заболеваний, лечение которых, согласно действующему законодательству, должно осуществляться за счет средств федерального и регионального бюджета.
Причины синдрома Хантера
Развитие синдрома Хантера связано с мутацией гена идуронатсульфатазы (IDS), кодирующего лизосомный фермент идуронат-2-сульфатазу. Ген IDS картирован в локусе Xq28 на длинном плече Х-хромосомы; в настоящее время известно более 150 различных его мутаций (точечные мутации, мелкие и крупные делеции, вставки, перестройки и пр.).
В силу сцепленности наследования с Х-хромосомой, синдромом Хантера, как правило, страдают исключительно мальчики (XY). Гетерозиготные женщины в подавляющем большинстве случаев являются носителями мукополисахаридоза II типа без клинических проявлений, однако описаны несколько случаев синдрома Хантера у девочек, связанных с новой мутацией или инактивацией второй, нормальной Х-хромосомы.
Мутации в гене IDS сопровождаются дефицитом или отсутствием фермента iduronate-2-sulfatase (I2S), неполным расщеплением и накоплением в лизосомах клеток практически всех тканей и органов гликозаминогликанов (ГАГ) – дерматансульфата и гепарансульфата.
Классификация синдрома Хантера
Степень выраженности клинических проявлений синдрома Хантера (среднетяжелая, тяжелая) зависит от типа мутации в гене IDS. Так, крупные делеции и сложные перестройки, как правило, обусловливают развитие тяжелых форм синдрома Хантера. Для тяжелой степени (мукополисахаридоза типа IIА) характерно ранее развитие клинических симптомов (преимущественно в возрасте 18-36 месяцев), быстро прогрессирующее течение, выраженная умственная отсталость, полиорганное поражение. Пациенты с тяжелой формой синдрома Хантера погибают на втором десятилетии жизни.
Среднетяжелая форма (мукополисахаридоз типа IIВ) составляет примерно 1/3 всех случаев патологии. Клинические проявления синдрома Хантера обычно возникают у детей в 3-8 (иногда 10-13) лет; интеллект обычно сохранен; продолжительность жизни при благоприятных условиях может достигать 50-60 лет. Пациенты с легкой формой синдрома Хантера могут успешно реализовать себя в профессиональной сфере и иметь здоровое потомство.
Симптомы синдрома Хантера
На момент рождения дети с синдромом Хантера выглядят клинически здоровыми. Основная симптоматика развивается в среднем в 2-4 года. До этого возраста проявления заболевания неспецифичны: у детей могут отмечаться повторные риниты, шумное дыхание, пупочные и паховые грыжи, гидроцеле.
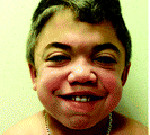
Ранним характерным признаком синдрома Хантера служит постепенное изменение внешности ребенка: черты лица становятся грубыми (гаргоилизм); язык, губы и ноздри – большими; кожа – толстой. Облик больного с синдромом Хантера дополняется низкорослостью, увеличением размеров головы (макроцефалией), короткой шеей, аномалиями зубных рядов (редкими зубами). Дети с синдромом Хантера внешне очень похожи друг на друга и напоминают братьев.
Другие ранние признаки мукополисахаридоза II типа включают хриплый низкий голос, увеличение живота, гепатоспленомегалию. Дети с синдромом Хантера склонны к частой заболеваемости ОРВИ, отитами, ларингитом, трахеитом, пневмониями; нередко у них отмечается обструктивное апноэ сна, хроническая диарея. Следствием отложения гликозаминогликанов и липидов в дерме служит возникновение узелково-папулезных высыпаний на коже плеч, бедер, лопаток. Возможно появление «монгольских пятен» в пояснично-крестцовой области, гипертрихоза.
Уже в дошкольном возрасте у детей с синдромом Хантера появляются признаки поражения опорно-двигательного аппарата, выражающиеся в тугоподвижности суставов, развитии кифосколиоза, деформации кистей по типу «когтистой лапы». Движения становятся затруднительными, походка неуклюжей; к юношескому возрасту больные с синдромом Хантера нередко делаются беспомощными инвалидами, прикованными к постели.
Неврологические нарушения, возникающие при синдроме Хантера, включают синдром гипервозбудимости, судороги, сообщающуюся гидроцефалию, спастическую параплегию, задержку речевого развития, прогрессирующую тугоухость. Со стороны зрительной системы обнаруживается помутнение роговицы, атипичный пигментный ретинит. К наиболее поздним признакам мукополисахаридоза II типа относятся кардиологические нарушения: появление шумов в сердце, приобретенные пороки сердца (чаще митральная недостаточность), кардиомиопатия и др.
У ребенка могут наблюдаться изменения психики: обидчивость, агрессивность. В зависимости от типа синдрома Хантера интеллектуальный дефект может быть небольшим или значительно выраженным. Летальный исход у больных с синдромом Хантера обычно наступает от прогрессирующей сердечной или легочной недостаточности.
Диагностика синдрома Хантера
В практике педиатра синдром Хантера встречается исключительно редко. Тем не менее, мукополисахаридоз II типа может быть заподозрен на основании клинических признаков (внешних изменений, манифестации заболевания на 2-4 году жизни, прогредиентного течения и др.). Для подтверждения диагноза дети нуждаются в консультации генетика, анализе клинико-генеалогических данных, проведении молекулярно-генетических исследований.
Важным биохимическим маркером синдрома Хантера служит повышение экскреции мукополисахаридов (дерматансульфата, гепарансульфата) с мочой, дефицит фермента идуронат-2-сульфатазы. Рентгенологические исследования костей черепа, суставов, трубчатых костей, позвоночника демонстрируют дизостоз, остеоартрит, множественные изменения позвонков.
Для обнаружения морфофункциональных изменений со стороны внутренних органов и ЦНС проводится УЗИ органов брюшной полости, ЭКГ, ЭхоКГ, электроэнцефалография, МРТ головного мозга. Морфологическое исследование тканей, полученных в результате биопсии кожи, печени, миокарда и пр., выявляет однотипные изменения – клетки, заполненные гликолипидами.
В семьях с известным генотипом и высоким риском рождения ребенка с синдромом Хантера может проводиться инвазивная пренатальная диагностика – биопсия ворсин хориона, амниоцентез или кордоцентез с определением активности идуронат-2-сульфатазы в полученном материале.
Дифференциальную диагностику синдрома Хантера следует проводить с другими формами мукополисахаридозов (прежде всего, синдромом Гурлера), а также с другими лизосомными болезнями накопления.
Лечение синдрома Хантера
Детям с синдрома Хантера необходимо пожизненное проведение ферментозаместительной терапии. На сегодняшний день единственным препаратом, зарегистрированным в США, Европе и России для лечения больных с синдромом Хантера, является идурсульфаза. Пациенты с мукополисахаридозом II типа должны получать идурсульфазу еженедельно путем внутривенного капельного введения в дозе 0,5 мг/кг массы тела.
Кроме патогенетического лечения, детям с синдромом Хантера проводятся регулярные медикаментозные курсы симптоматической и корригирующей терапии гепатопротекторами, витаминами, антиоксидантами, цитопротекторами. В комплексную терапию синдрома Хантера целесообразно включать ЛФК, физиотерапию (электрофорез лидазы на суставы, парафиновые аппликации, магнитотерапию, лазеропунктуру), занятия с дефектологом и логопедом.
Прогноз и профилактика синдрома Хантера
Синдром Хантера типа В имеет более благоприятное течение и прогноз; при своевременном и регулярном лечении продолжительность жизни больных может достигать 50-60 лет. При тяжелых формах мукополисахаридоза II типа пациенты обычно погибают до 20 лет от сердечно-сосудистой недостаточности. На сегодняшний день серьезную проблему для больных с синдромом Хантера представляет своевременное получение жизненно необходимого препарата идурсульфазы из-за его высокой стоимости.
Дети с синдромом Хантера нуждаются в наблюдении различных специалистов: детского генетика, педиатра, детского кардиолога, детского невролога, эпилептолога, детского офтальмолога, детского отоларинголога, детского хирурга, детского ортопеда.
Основным методом профилактики синдрома Хантера является медико-генетическое консультирование супружеских пар, имеющих вероятность рождения больного ребенка, а также проведение дородовой диагностики. Следует знать, что у больных с синдромом Хантера рождаются здоровые сыновья, а дочери выступают облигантыми носителями мутантного гена.
Источник: https://www.krasotaimedicina.ru/diseases/children/hunter-syndrome





